Helix’s Exome+® assay is run on all samples sequenced at Helix. This single assay combines a clinical exome with uniform panel-grade tests, a genome-wide backbone, and full mitochondrial coverage. Post sequencing, rigorous quality checks are applied to ensure that the data generated are appropriate for a broad application of clinical genetic tests. These tests range from population health screens, to diagnostic or pharmacogenomic panels to whole exome sequencing. Meanwhile, the Exome+ backbone of whole genome single nucleotide polymorphisms (SNPs), augmented with imputation, enables the generation of ancestry, traits, and polygenic risk scores. By ensuring that this single assay is appropriate for answering a wide list of clinical questions, the Exome+ assay establishes a foundation for Sequence Once, Query Often® . Sequence Once, Query Often is a process that allows various genetic test results to be delivered for an individual over the course of their life based on when each might be relevant or informative.
In building a test menu relying on the Exome+ assay, we have focused on three important pillars: quality, breadth of testing, and turnaround time, as discussed in the following sections.
Quality of Testing
Coverage and Callability
Over the past decade, Helix has optimized the Exome+ assay for uniformity of coverage, and target region completeness in order to maximize scalability and utility of genetic testing. Our assay focuses on callability (percent of targeted bases that produce a reliable genotype call) and CNV sensitivity by balancing depth of coverage & mapping/alignment quality metrics down to the individual base level . For all diagnostic panels, we guarantee a callability rate exceeding 99.8%. Performance is delivered efficiently through a tiered coverage approach, resulting in ~140x coverage across cancer genes, ~85x coverage across cardio genes, and > 500x coverage across the mitochondrial genome.
Variant Calling
Small variants and copy number variants (CNVs) are generated across the full exome, with performance of these variants captured in Table 1. Small variants are called using a customized version of Sentieon while all other variant types are called using proprietary algorithms. For example, CNVs are detected by both read depth and split reads. For variants that are more challenging— for example, due to interference by pseudogenes (e.g. PMS2 terminal exons) or special variant types (e.g. the MSH2 inversion also known as the Boland Inversion)— specialized informatics are applied and a special validation is performed.
Small variant results are based on whole-exome data from five National Institute of Standards and Technology (NIST) cell lines. CNV results are based on 36 known single-exon CNVs and 73 known multi-exon CNVs. The Boland inversion detection rate is measured against 3 samples run in duplicate. The BRCA2 Alu insertion detection rate is measured against 3 samples across 11 replicates. Detection rates for PMS2 small variants were measured against 13 samples across 45 replicates. Detection rates for PMS2 CNVs were measured against 24 samples across 40 replicates.
Table 1: Summary performance metrics for variants on the Helix Exome+ assay based on repeated analytical validations.
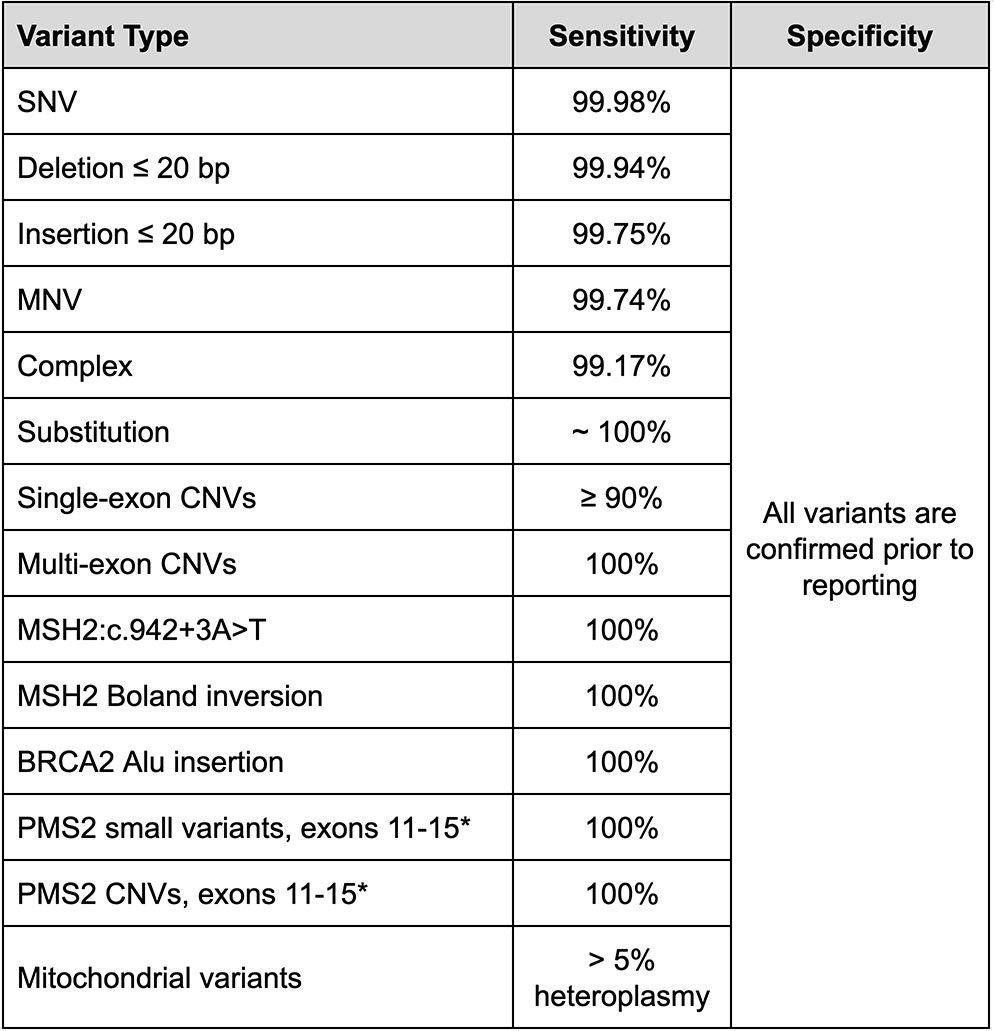
SNV = Single nucleotide variants. MNV = multi-nucleotide variant, or phased variants. Substitutions = an insertion and deletion with different lengths sharing the same location and strand. Complex = two different variant types sharing the same location but mapping to a different allele.
* PMS2 terminal exons are included in all diagnostic tests but not included in the Tier One Population Screen and other screening tests.
Additionally, we conducted validation studies to compare Exome+ small variants against Sanger sequencing. Analysis included 1,236 samples (1,141 sourced from saliva and 95 from cell lines) with 1,251 variants and 172,711 reference sites, and demonstrated > 99.9% concordance between Exome+ and Sanger.
Variant Interpretation
Variant interpretation is performed by scientists with deep expertise in human molecular genetics, following the guidelines established by the American College of Medical Genetics and Genomics (ACMG). To support this process, we have developed Helix Wayfinder™, a proprietary AI-powered tool that gathers and synthesizes information from relevant and trusted resources — including PubMed, ClinVar, gnomAD, pathogenicity prediction tools such as REVEL scores, and more — to ensure that scientists have the most up-to-date and complete picture of evidence available for each variant.
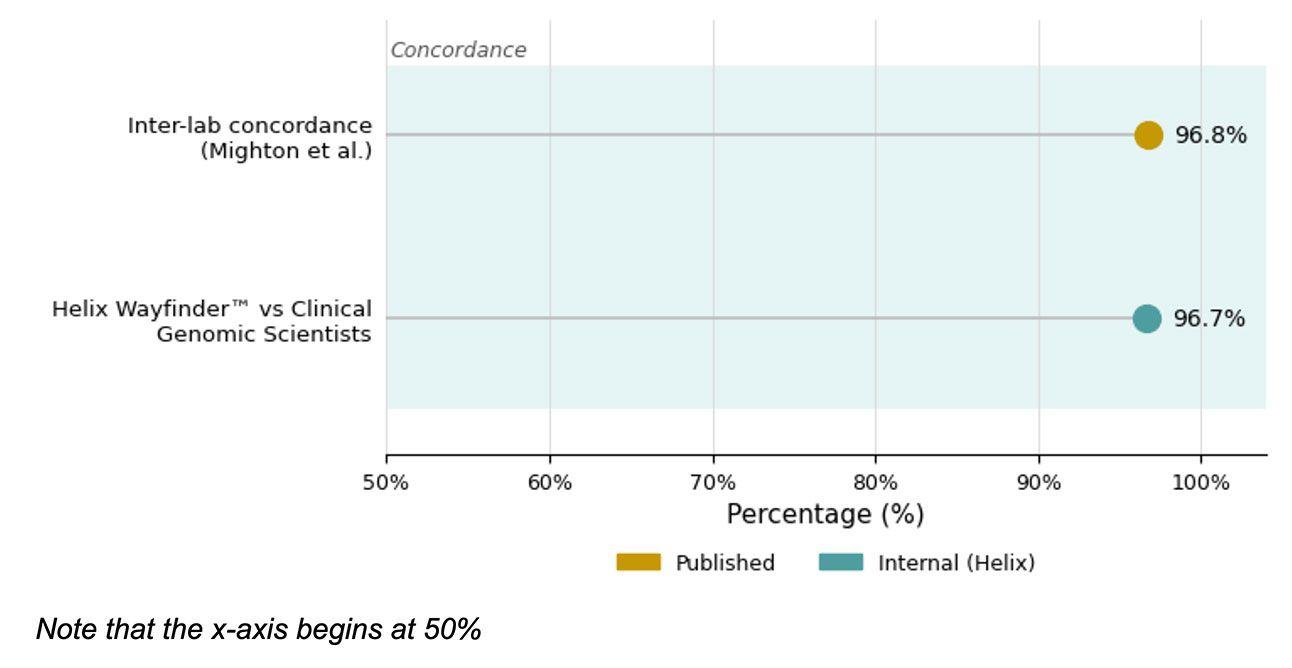
In a preliminary evaluation of Helix Wayfinder™ performance, variant classifications were assessed using a 2-tier concordance framework in which variants are grouped as either clinically actionable (Pathogenic and Likely Pathogenic) or not clinically actionable (Benign, Likely Benign, and Variants of Uncertain Significance). Even without additional input from clinical genomic scientists, Helix Wayfinder™ alone achieved >96.6% concordance (116/120) with manually verified classifications from clinical genomic scientists (Figure 1). To contextualize these findings, a multi-laboratory concordance study² found that prior to laboratory discussions to harmonize classifications, 2-tier concordance among clinical genomic scientists across laboratories was 96.8% — comparable to Helix Wayfinder™ performance. These results underscore Helix Wayfinder™ reliability as a decision-support tool, performing on par with the level of agreement observed among human experts across clinical laboratories.
Figure 1: Summary performance metrics for variants evaluated with Helix Wayfinder
Variant Confirmation
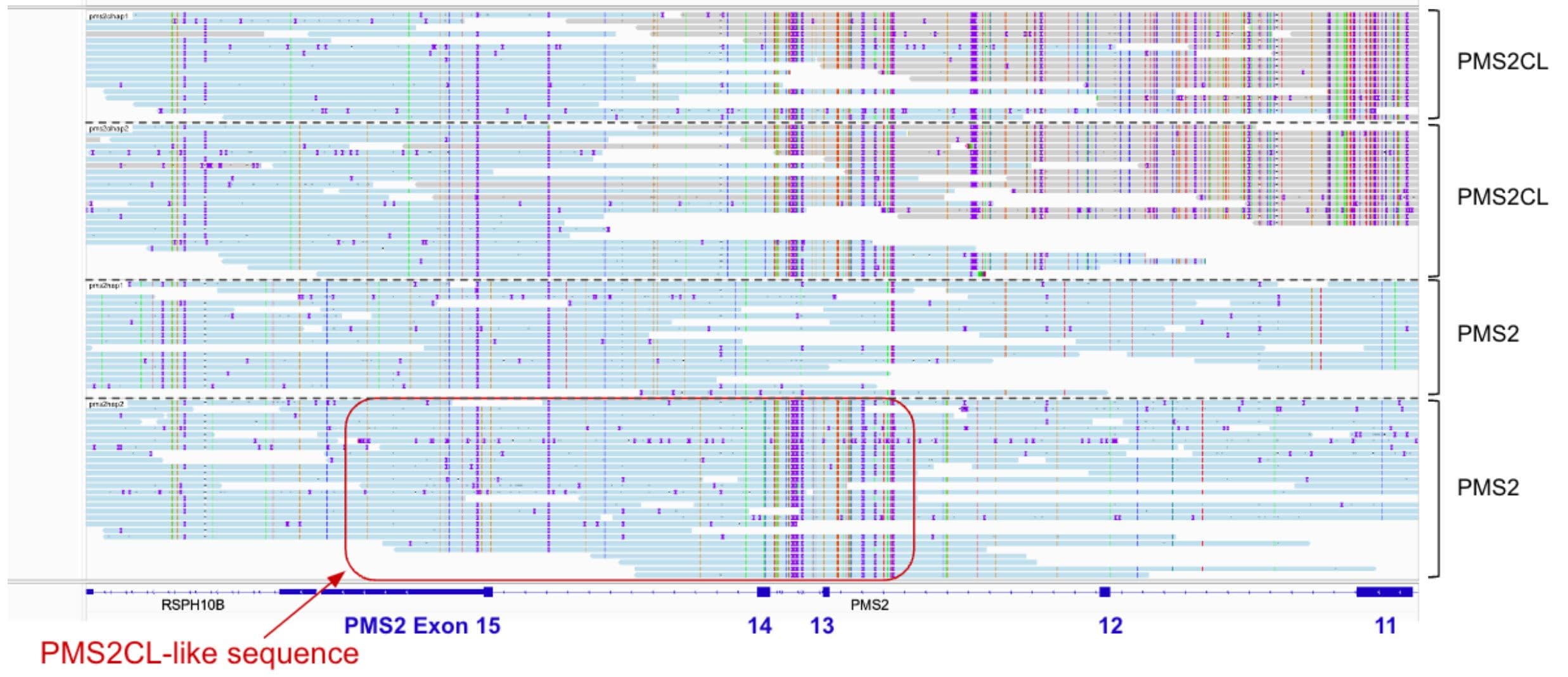
All variants are confirmed before reporting. Confirmation methods include visual inspection of the reads in simple cases such as SNVs with clear allele fractions and no concerning genomic context, short read whole genome sequencing for more complicated small variants and CNVs, and long read sequencing when appropriate. All PMS2 variants in terminal exons are confirmed by PacBio long reads. This allows us to confidently determine which variants are in PMS2 and which are in PMS2CL, and also to identify gene conversion events such as the one seen in Figure 2.
Figure 2: Screenshot of a PacBio run in which a saliva sample has been flagged for confirmation using the Exome+ assay. A gene conversion event was identified in a saliva sample using PacBio long-read sequencing. In the second PMS2 allele, exons 1-12 were present as usual, but were combined with PMS2CL exons 4-6 in place of PMS2 exons 13-15.
Pharmacogenomics
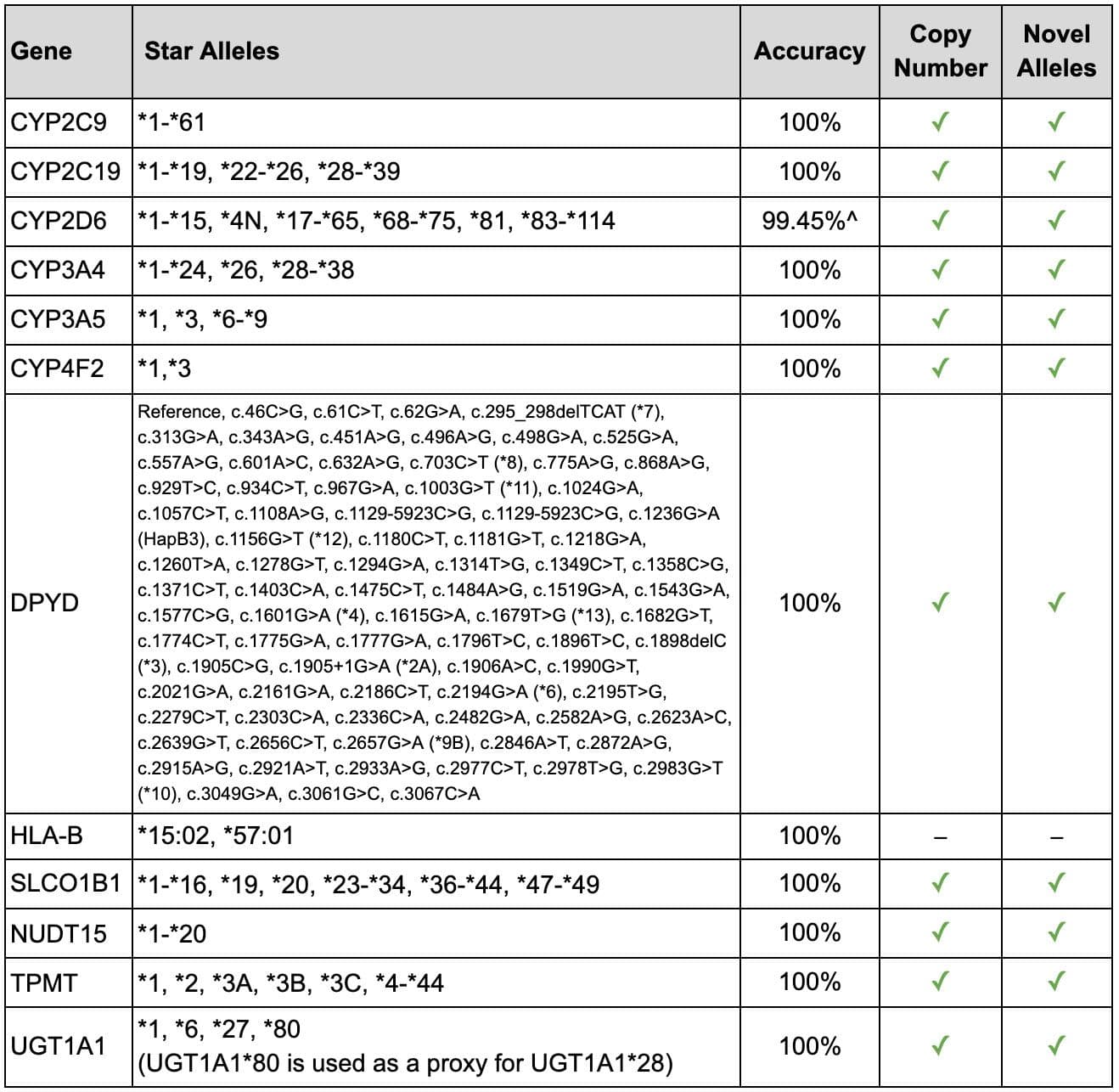
Whole exome sequencing is performed on every sample received for clinical genetic testing. Therefore, pharmacogenomic testing is based on whole gene results and not limited to targeted star alleles. For all genes with star alleles, all known star alleles are determined regardless of how common or rare they may be, and novel alleles are flagged. This ensures that the most accurate metabolizer status can be reported. Additionally, special informatics have been applied to CYP2D6 to ensure the reliable detection of star alleles representing gene fusions and copy numbers up to 5 copies. Both accuracy and range of results for pharmacogenomic genes is captured in Table 2, with results based on > 300 data sets from Genetic Testing Reference Material Coordination Program (GeT-RM) samples.
Table 2: Summary of reportable and analytical range and accuracy for Pharmacogenomic genes with star alleles on the Exome+ assay. Not listed, but included in our tests, are individual SNPs such as for VKORC1.
^ Two incorrect calls were identified in samples with a complex CYP2D6 rearrangement, as follows:
1. Truth = *1/*10/*36/*36; Call = *1/*10/*10/*36, both are Normal Metabolizers
2. Truth = *10/*36/*36; Call = *10/*10/*36, both are Intermediate Metabolizers
Comprehensive PGx tests are necessary to avoid misclassifications
Comprehensive PGx tests are necessary to avoid misclassifications
Helix’s approach to a comprehensive reportable range for pharmacogenomics is critical to supporting diverse populations. When star alleles are excluded from the reportable range, approximately 16.8% individuals are often mis-classified as *1, resulting in a normal metabolizer classification when the metabolizer status is in fact unknown (Table 3)¹. Additionally, gene copy number is important to understanding true metabolizer status, as duplications can quickly increase metabolism. Across a set of 16k individuals, ~ 16% of samples were identified to have whole gene CYP2D6 duplications (with another 5% harboring whole gene deletions), highlighting the importance of analyzing CYP2D6 copy number in conjunction with CYP2D6 star alleles.
Table 3: Impact of using an incomplete CYP2D6 panel. This table presents the outcome of using CYP2D6 tests with an incomplete star allele list as compared to the Helix PGx Pipeline, based on 30,000 individuals. For comparison, a representative panel was used that includes the common alleles: *1, *2, *3, *4, *4N, *5, *6, *9, *10, *17, *29, *35, *36, *41, and duplications.
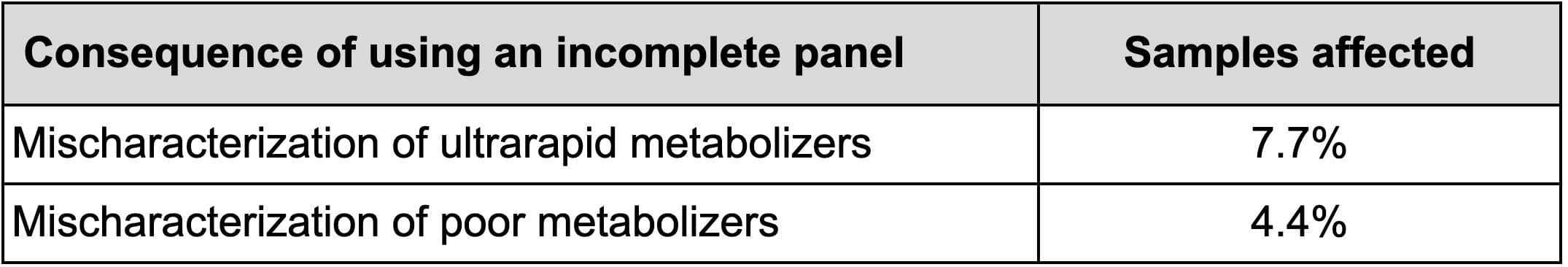
Helix reports all star alleles within the reportable range and includes assessment and reporting of gene copy number and novel star alleles to ensure the most accurate results for all populations.
Imputation
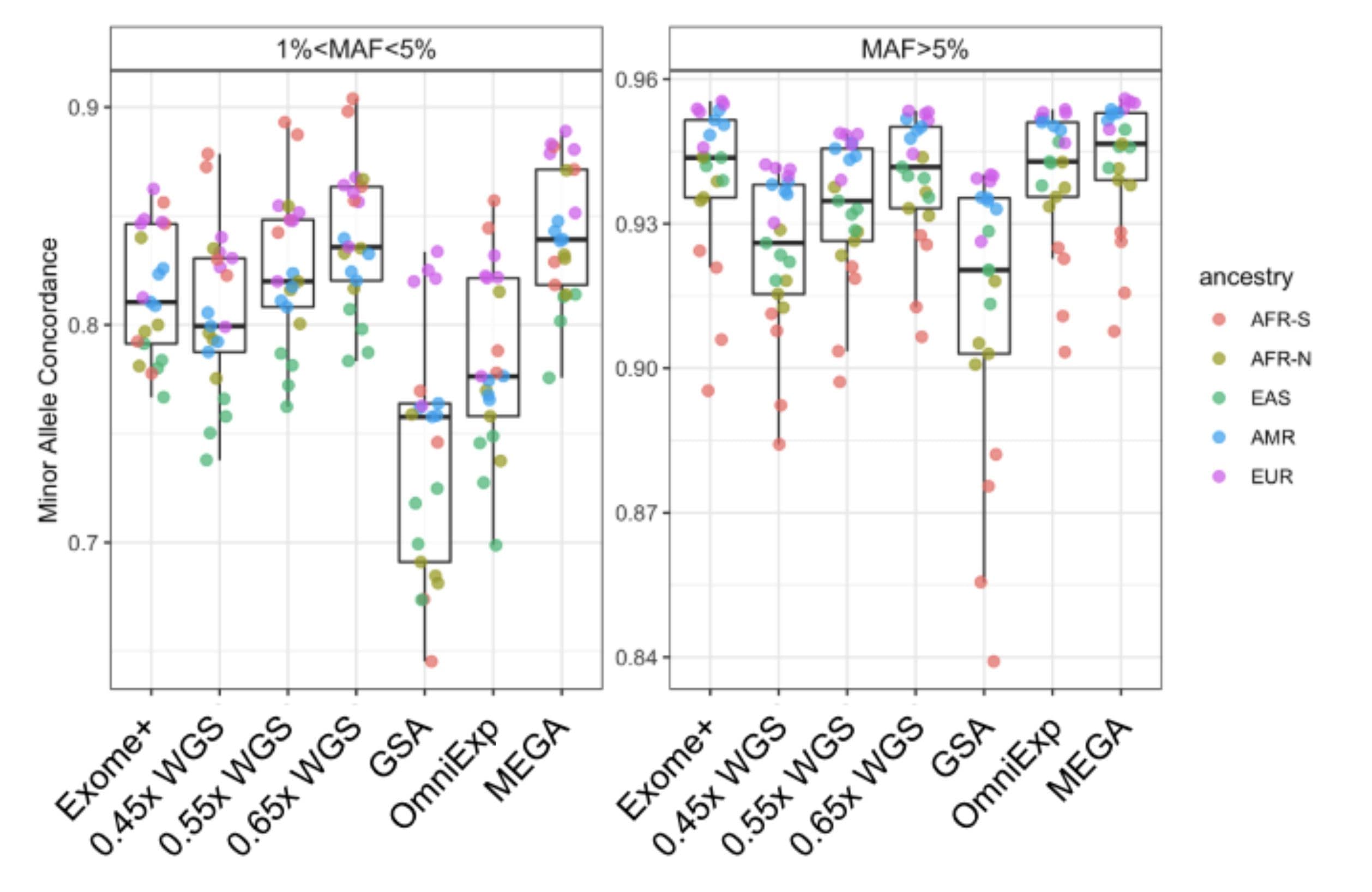
Both ancestry and polygenic risk scores can be easily and automatically generated from Exome+ results, leveraging a backbone of SNPs across the full genome that complements the exome itself. As a result, Helix is able to offer robust genome-wide imputation with tens of millions of high-confidence SNPs imputed with technical equivalence to ~0.6x WGS, Infinium OmniExpress, and Infinium MEGA for common alleles (Figure 3). For rare alleles, the Exome+ results were approximately equivalent to 0.5x WGS, with improved performance over GSA and OmniExp (Figure 3). This strong performance is possible because imputation is not limited to the hundreds of thousands of SNPs targeted in the Exome+ assay, but instead leverages all directly sequenced data, including the flanking regions of those SNPs and the full exome.
Figure 3: Demonstration of minor allele concordance between imputed variants and directly sequenced variants across five ancestral populations. Results are presented for typical Exome+ runs, WGS at different coverage levels (0.45x, 0.55x, 0.65x), and three microarrays (GSA, OmniExp, and MEGA). AFR-S: Sub-saharan African. AFR-N: North African. EAS: East Asian. AMR: Indigenous American. EUR: European.
Breadth of Testing
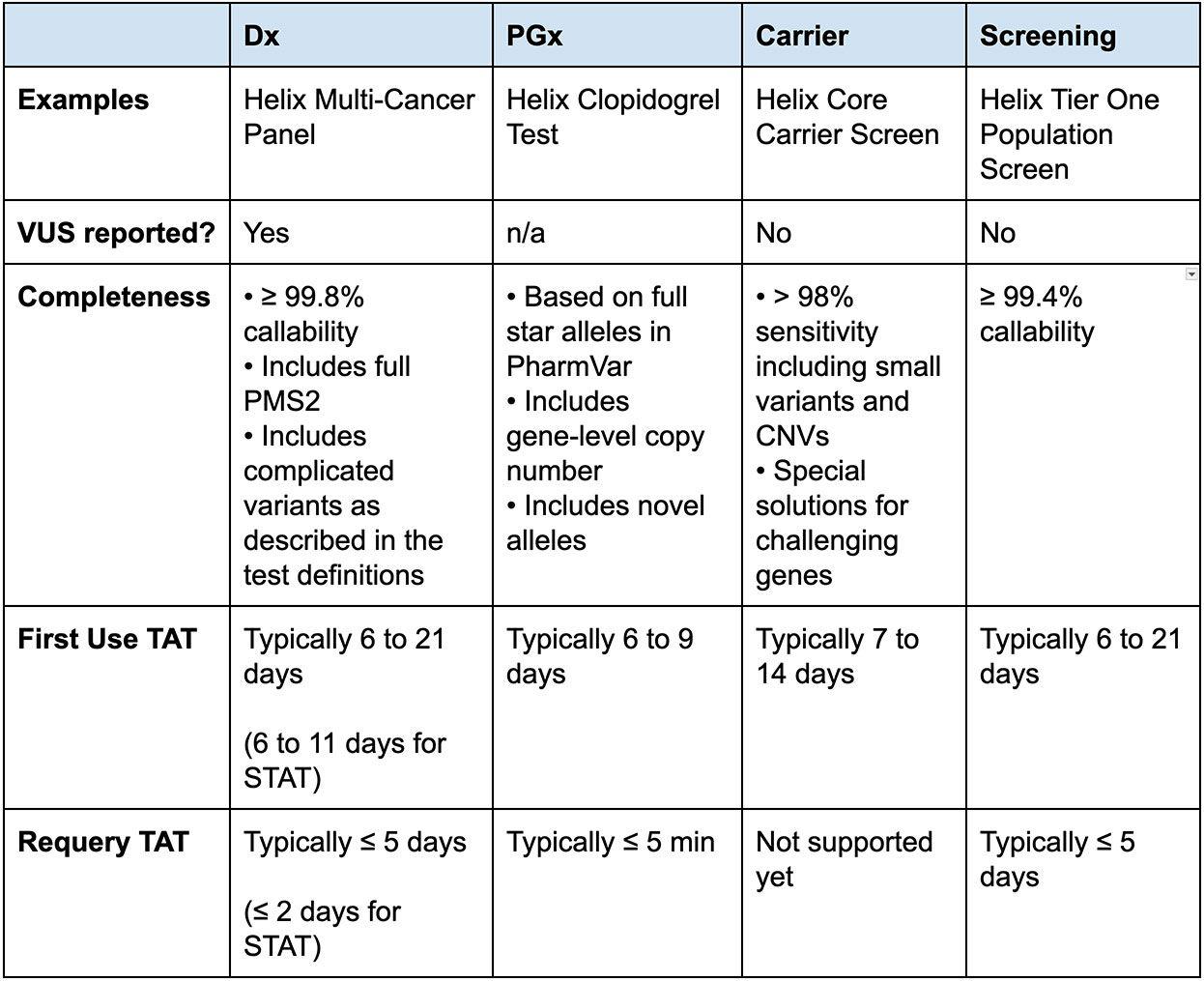
Helix’s Exome+ assay has been designed to be used for a wide range of testing categories and additional informatic tools or procedures to ensure fast turnaround times are implemented according to the tests’ specific needs (Table 4). This enables Helix to offer a variety of high-quality efficient testing options based on a single sample and sequencing.
Table 4: Comparison of Helix Test Types and Characteristics
Polygenic Risk Scores
In addition to developing panel-grade products, we also have the capability to impute tens of millions of common genotypes in support of polygenic risk score applications, which are intended to be added as complements to our diagnostic tests in the near future. Polygenic Risk Scores (PRS) are emerging as a powerful tool to predict an individual’s risk of disease. PRS can be used to predict disease risk as effectively as variants within single genes.
Because PRS are based on many genomic loci, often focused in non-coding regions, their coverage requirements are significantly different from those for standard genetic testing of monogenic disease. The Exome+ assay supports analysis of PRS with direct coverage of hundreds of thousands of non-coding variants plus tens of millions of genome-wide imputed variants. These millions of variant calls ensure that PRS of all sizes are supported.
Ancestry
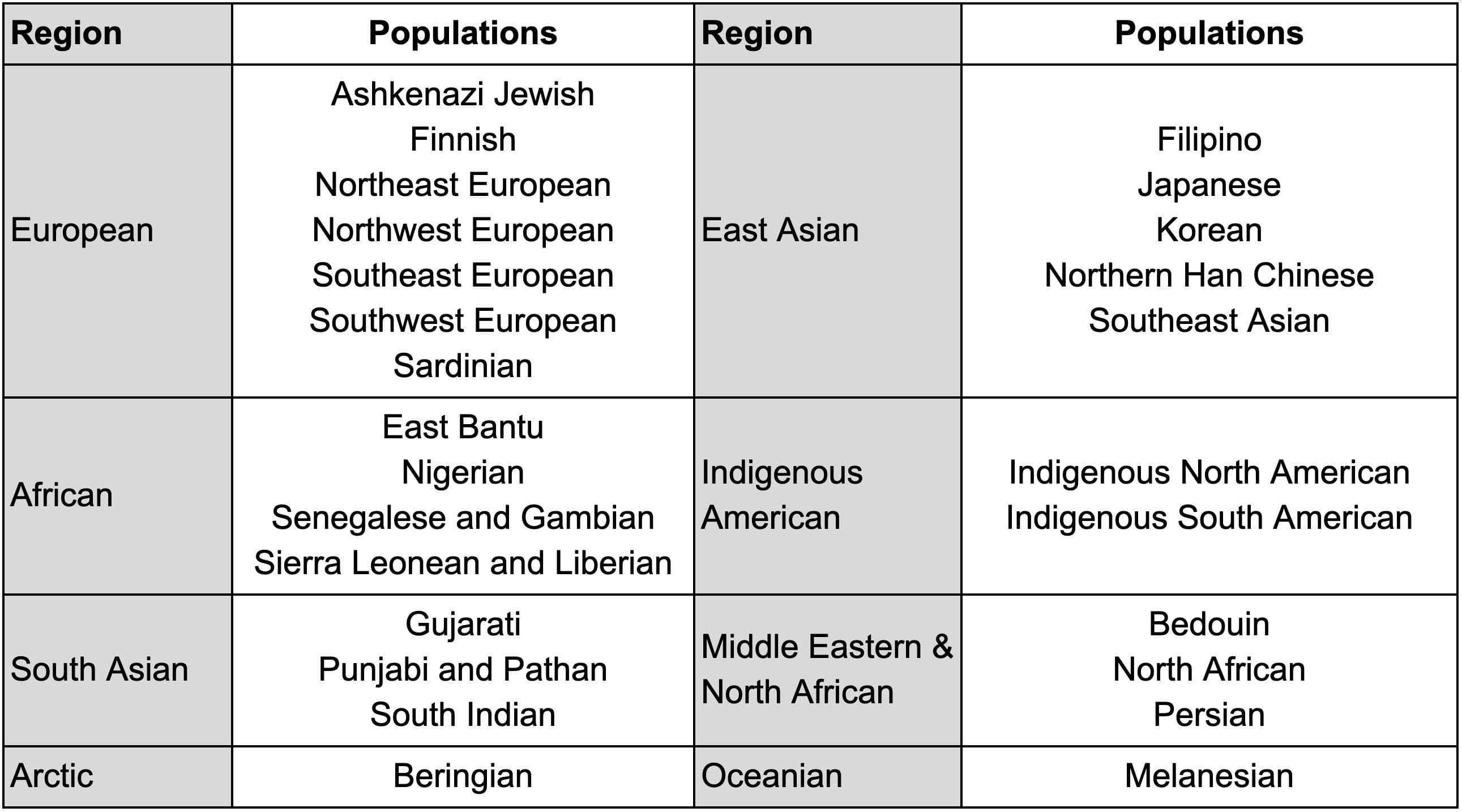
Ancestry can help provide context for the relevance of clinical genetic testing, for example when certain populations have an increased concentration of carriers of specific risk alleles. The Exome+ assay results in ancestry assignments spanning 26 ancestries across eight regions (Table 5).
Table 5: List of 26 populations, grouped into eight regions, calculated by Helix.
Turnaround time
The Exome+ assay offers the basis for Sequence Once, Query Often processes, where the data generated upon initial testing can be used during additional queries. The turnaround time for Helix tests will be significantly reduced for patients who have already been sequenced at Helix. Table 4 summarizes typical turnaround time based on different test types.
Diagnostic and pharmacogenomic tests are prioritized within the Helix lab in order to meet quick turnaround times. After a provider orders a genetic test, Helix performs whole exome sequencing and then interprets only the specific genes within the test ordered. Helix then continues to store the patient’s genetic information for future clinical use. With the patient’s permission, their health care provider can order future medically necessary genetic tests from Helix without the need to submit another sample (in most cases). Tests that can be fully automated through digital analysis of a patient’s genetic information, such as PGx tests, can typically be returned in under five minutes.
References
- A comprehensive landscape of CYP2D6 variation across 30,000 individuals
- Mighton C, Smith AC, Mayers J, Tomaszewski R, Taylor S, Hume S, Agatep R, Spriggs E, Feilotter HE, Semenuk L, Wong H, Lazo de la Vega L, Marshall CR, Axford MM, Silver T, Charames GS, Di Gioacchino V, Watkins N, Foulkes WD, Clavier M, Hamel N, Chong G, Lamont RE, Parboosingh J, Karsan A, Bosdet I, Young SS, Tucker T, Akbari MR, Speevak MD, Vaags AK, Lebo MS, Lerner-Ellis J; Canadian Open Genetics Repository Working Group. Data sharing to improve concordance in variant interpretation across laboratories: results from the Canadian Open Genetics Repository. J Med Genet. 2022 Jun;59(6):571-578. doi: 10.1136/jmedgenet-2021-107738. Epub 2021 Apr 19. PMID: 33875564; PMCID: PMC8523590.
Updated March 2026. The Exome+® assay is run exclusively at Helix’s CLIA and CAP accredited laboratory facility in San Diego, CA (CLIA #05D2117342, CAP #9382893). The Helix Laboratory is a highly automated facility with the ability to process millions of Exome+ assays annually.