Highlights
- We report on sixteen SARS-CoV-2 sequences harboring both E484Q and L452R substitutions, the characteristic mutations in the B.1.617 “double mutant” lineage.
- Two of these sixteen samples belong to lineage B.1.429, which, up to now, has not been associated with the E484Q substitution.
- Both E484Q and L452R have been shown to help the virus avoid the immune system’s antibody response; their emergence in separate viral lineages suggests convergent evolution in response to selective pressure.
- The prevention of a variants-driven surge in the United States has focused on limiting travel from countries reporting a high prevalence of these variants, such as India. However, our findings suggest that lineages of the SARS-CoV-2 virus that are already circulating in the United States are under selective pressure and may well develop similar variants that help them avoid the immune system, rendering current mitigation efforts less effective.
Introduction
On March 24, 2021, the Indian Ministry of Health and Family Welfare announced the identification of a potential new lineage of the SARS-CoV-2 virus that harbors two substitutions, E484Q and L452R, that “confer immune escape and increased infectivity” 1. This lineage, which at the time was estimated to be responsible for 15-20% of all infections in India, was later designated as the B.1.617 lineage after evidence of its spread to the UK, Australia, New Zealand, Singapore, USA, Germany, and Canada came to light2. It has also been dubbed by some media outlets as the “India variant”, for the geographical region it was first reported, or “double mutant”, for the two key amino acid substitutions that it harbors (E484Q and L452R)3.
A note on Variant vs Lineages
The terms “variant” and “lineage” have been used interchangeably in broader discussions about the SARS-CoV-2 virus’ evolution. These terms are typically used to refer to a new version of the SARS-CoV-2 virus and to highlight its relationship to past or future versions. Here, in a bid for clarity and consistency, we’ve opted to primarily use the term “lineage” when referencing discrete, genetically identifiable versions of the SARS-CoV-2 virus, such as B.1.1.7 (initially known as the “UK Variant”) and B.1.617 (broadly known as the “double mutant variant”).
E484K and L452R are considered “SARS-CoV-2 Substitutions of Therapeutic Concern” by the CDC4. Substitutions in both of these positions on the Spike protein have been shown to convey immune escape against human monoclonal antibodies as well as convalescent human sera—meaning these mutations can diminish the body’s ability to recognize and respond to the virus5,6. While E484Q is not the same substitution as E484K, it shares the same functional outcome: it enables the virus to escape monoclonal antibodies5. E484K is present in multiple lineages of concern (also known as variants of concern), such as P.1, B.1.351, and some sequences of B.1.1.7. Similarly, the L452R substitution is found in sequences belonging to the B.1.427 and B.1.429 lineages. Thus, the presence of both these spike protein substitutions in the same viral sequence potentially signals a developing SARS-CoV-2 lineage that is better able to escape immunity or to have increased infectivity compared to those that are already present.
Results
Here, we report on sixteen whole-genome SARS-CoV-2 sequences in our database that carry both L452R and E484Q mutations (Table 1). While thirteen of these were designated B.1.617, as expected, two of the nine belonged to the B.1.429 lineage, which has so far not been associated with either E484Q or E484K. (The one other L452R+E484Q sample was designated B.1.609, which is neither a variant of concern nor interest.)
We confirmed that the two B.1.429 sequences did indeed have strong read support for the two mutations (more than 500 reads support the alternative allele in both positions in both samples). We also confirmed that the two sequences carried the other spike substitutions typically associated with the B.1.429 lineage (S13I and W152C).
Table 1: Attributes of the L452R+E484Q carriers and three comparison sequences▼
| GISAID virus name | Collection date | Pango lineage | Escape muts S:E484Q | Escape muts S:L452R | Other B.1.429 muts: S:S131 | Other B.1.429 muts S:W152C |
|---|---|---|---|---|---|---|
| hCoV-19/USA/CA-CDC-STM-000022631/2021 | 2021-02-24 | B.1.429 | Y | Y | Y | Y |
| hCoV-19/USA/CA-CDC-STM-000027958/2021 | 2021-03-01 | B.1.429 | Y | Y | Y | Y |
| hCoV-19/USA/PA-CDC-STM-000052931/2021 | 2021-04-08 | B.1.609 | Y | Y | N | N |
| hCoV-19/USA/FL-CDC-STM-000028344/2021 | 2021-03-03 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/GA-CDC-STM-000041012/2021 | 2021-03-21 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/GA-CDC-STM-000042157/2021 | 2021-03-22 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/CA-CDC-STM-000042578/2021 | 2021-03-24 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/PA-CDC-STM-000043645/2021 | 2021-03-24 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/GA-CDC-STM-000046368/2021 | 2021-03-31 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/FL-CDC-STM-000049137/2021 | 2021-04-01 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/FL-CDC-STM-000053252/2021 | 2021-04-09 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/CA-CDC-STM-000054394/2021 | 2021-04-11 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/IL-CDC-STM-000053846/2021 | 2021-04-11 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/IL-CDC-STM-000056003/2021 | 2021-04-13 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/IN-CDC-STM-000056444/2021 | 2021-04-14 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/MI-CDC-STM-000056932/2021 | 2021-04-15 | B.1.617 | Y | Y | N | N |
| hCoV-19/India/WB-1931300243737/2021* | 2021-03-11 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/CA-Stanford-19_S07/2021** | 2021-04-06 | B.1.617 | Y | Y | N | N |
| hCoV-19/USA/CA-Stanford-19_S21/2021** | 2021-04-07 | B.1.617 | Y | Y | N | N |
+: More recent sequences are currently in the process of being submitted to GISAID.
*: Sequence initially submitted to support the new B.1.617 lineage designation.
**: Two sequences identified in California that were of the B.1.617 lineage.
As an additional check, since both of the B.1.429 sequences with L452R+E484Q were collected in California, we investigated whether these two sequences were phylogenetically related to two other California samples that were independently sequenced and designated as B.1.6177. We used Nextclade8 to place all these sequences in a phylogenetic tree representative of the global diversity of SARS-CoV-2 (Figure 1).
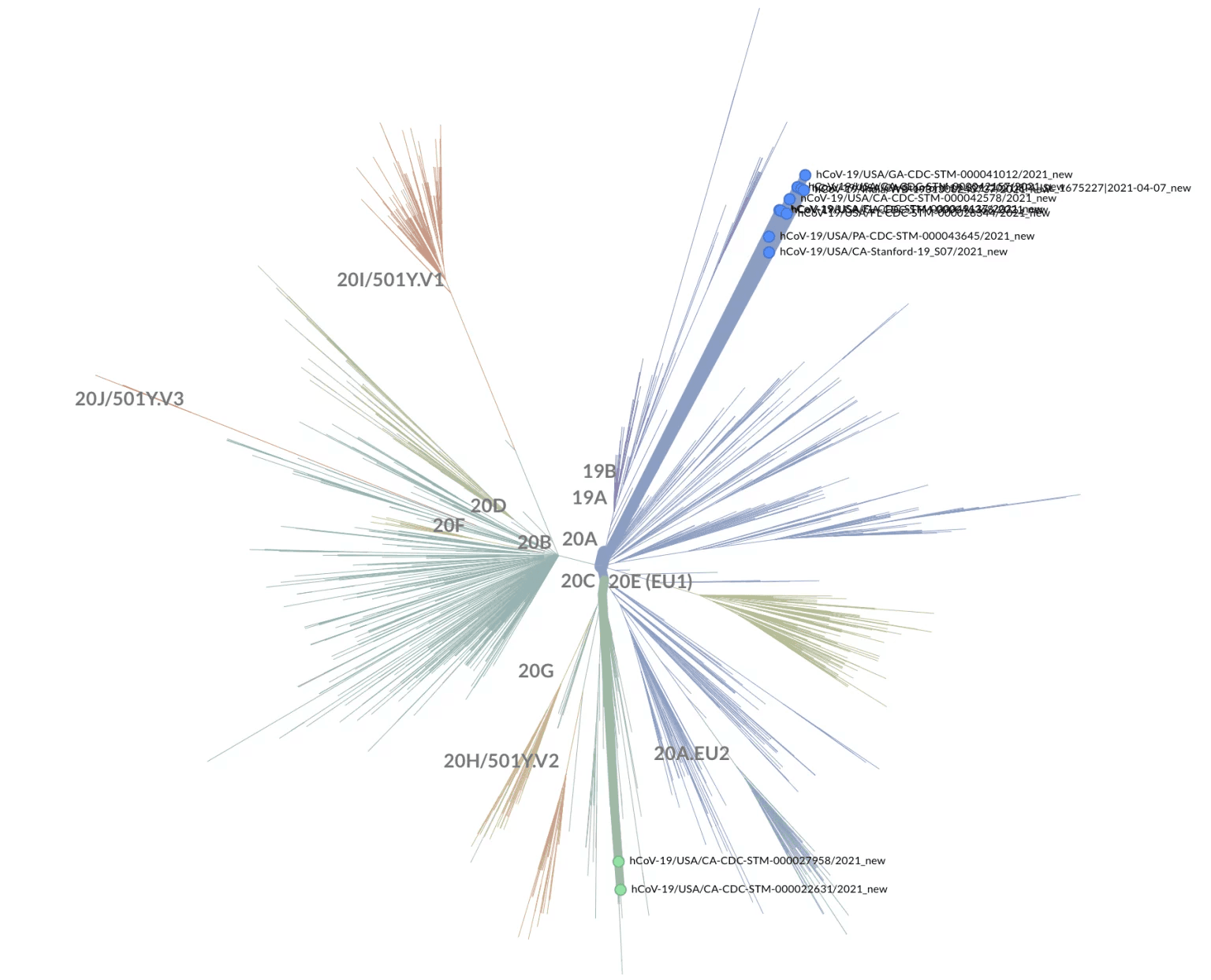
Figure 1: Phylogenetic tree with subset of L452R+E484Q samples in Table 1 on background of global SARS-CoV-2 diversity. Note that the two B.1.429 samples with L452R+E484Q are in a different nextclade clade (20C, green) from the B.1.617 samples and two independently sequenced California B.1.617 samples (20A, blue). Generated using https://clades.nextstrain.org/ and selecting “Show Tree” > “Unrooted” layout.
As is clear from both Table 1 and Figure 1, the two B.1.429 samples with the L452R+E484Q substitutions are part of a separate SARS-CoV-2 evolutionary lineage from the other L452R+E484Q samples. That the L452R+E484Q genotype is, or has been, present in California in both B.1.617 and B.1.429 lineages suggests that the evolution of the SARS-CoV-2 genome might be driven by convergence to substitutions that convey a selective advantage.
Put another way, the presence of these mutations in two separate lineages suggests that they evolved separately, likely because mutations in these locations help the virus survive and outperform other lineages. It would be expected, then, that mutations of this sort (substitutions at L452 or E484, referred to as S:L452 and S:E484) will continue to develop independently in existing lineages and give rise to new variant lineages that are better able to avoid the immune system.
Indeed, when we extend our investigation to all substitutions in the S:L452 and S:E484 residues, we find that multiple SARS-CoV-2 lineages in the United States have chanced upon double mutations in these positions (Table 2). In addition to the S:E484Q substitution on the B.1.429 background above, there are also two instances of a S:E484K substitution on a B.1.429 background, as well as six instances of a S:E484G substitution on a B.1.526.1 background.
Table 2: All instances of double substitutions in the S:L452 and S:E484 residues▼
| S:L452* | S:E484 | Pango lineage | Count | States | Earliest date | Most recent date |
|---|---|---|---|---|---|---|
| S:E484G | S:L452R | B.1.526.1 | 6 | GA, IN | 2021-02-16 | 2021-03-06 |
| S:E484K | S:L452R | B.1.429 | 2 | TX | 2021-02-21 | 2021-02-21 |
| S:E484Q | S:L452R | B.1.429 | 2 | CA | 2021-02-24 | 2021-03-01 |
| S:E484Q | S:L452R | B.1.609 | 1 | PA, FL, GA, CA | 2021-04-08 | 2021-04-08 |
| S:E484Q | S:L452R | B.1.617 | 13 | PA, IL, IN, MI | 2021-03-03 | 2021-04-15 |
* No substitutions, other than S:L452R, were found in combination with S:E484 substitutions.
Given that the current dominant SARS-CoV-2 lineage, B.1.1.7, does not always carry substitutions in the 452 and 484 positions of the spike protein4, and given that the number of new daily cases have either been flat or declining in the past month, the immediate risk of a surge in L452R+E484Q variants due to convergent evolution in the United States is low. Imported B.1.617 strains are currently the primary source of these “double mutant” variants (representing 13 out of 24 instances in Table 2). Nonetheless, our findings here suggest that domestically circulating SARS-CoV-2 lineages already appear primed to develop similar mutations that might give rise to lineages of the SARS-CoV-2 virus that ultimately render our current mitigation efforts ineffective.
Methods
With the exception of the B.1.617 samples hCoV-19/India/WB-1931300243737/2021, hCoV-19/USA/CA-Stanford-19_S07/2021, and hCoV-19/USA/CA-Stanford-19_S21/2021, all viral samples reported here were collected by Helix through its COVID-19 diagnostic testing lab. Sequencing was carried out in partnership with Illumina as part of the SARS-CoV-2 genomic surveillance program led by the Centers for Disease Control and Prevention (CDC). To date, Helix has contributed over 46,000 samples towards this effort.
For each sample’s consensus sequence, the Pango lineage9,10 was obtained by running pangolearn version 2021-04-01 (https://github.com/cov-lineages/pangoLEARN) and the annotation of amino acid changes was obtained by running nextclade version 0.14.2 (https://github.com/nextstrain/nextclade).
Data reported here are current as of May 3, 2021 and may not reflect more recent sequencing results.
References
- Genome Sequencing by INSACOG shows variants of concern and a Novel variant in India. https://pib.gov.in/PressReleaseIframePage.aspx?PRID=1707177.
- pango-designation. (Github).
- BBC News. Coronavirus: ‘Double mutant’ Covid variant found in India. BBC (2021).
- CDC. Cases, Data, and Surveillance. https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html (2021).
- Greaney, A. J. et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 29, 44–57.e9 (2021).
- Liu, Z. et al. Landscape analysis of escape variants identifies SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. bioRxiv (2020) doi:10.1101/2020.11.06.372037.
- WFTV. https://www.wftv.com/news/trending/coronavirus-new-covid-19-variant-first-detected-[…]india-found-san-francisco-bay-area/6OWQFI2JIBBFFPGMTB2ESIS76I/.
- Nextclade. https://clades.nextstrain.org/.
- Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 5, 1403–1407 (2020).
- Rambaut, A. et al. Addendum: A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nature microbiology vol. 6 415 (2021).