This blog described findings currently published on bioRxiv.
You can read the pre-print article here >
In recent months, our world has been hit by the novel coronavirus SARS-CoV-2, which causes the disease COVID-19. Multiple groups around the world, including Helix, are currently working to genotype and/or sequence individuals who have been infected with SARS-CoV-2, and make their datasets available, in order to study potential host genetics and susceptibility factors for COVID-191. During the HIV epidemic, researchers were able to discover that approximately 1% of European ancestry individuals were immune to HIV infection due to a deletion in the host coreceptor CCR52–4. Similar genetic findings could help us identify individuals who are not at risk for COVID-19 or who are predisposed to certain disease outcomes. While no one has yet sequenced infected individuals, we are able to examine the naturally occurring variation at the population level in genes suspected to play a role in SARS-CoV-2 infection.
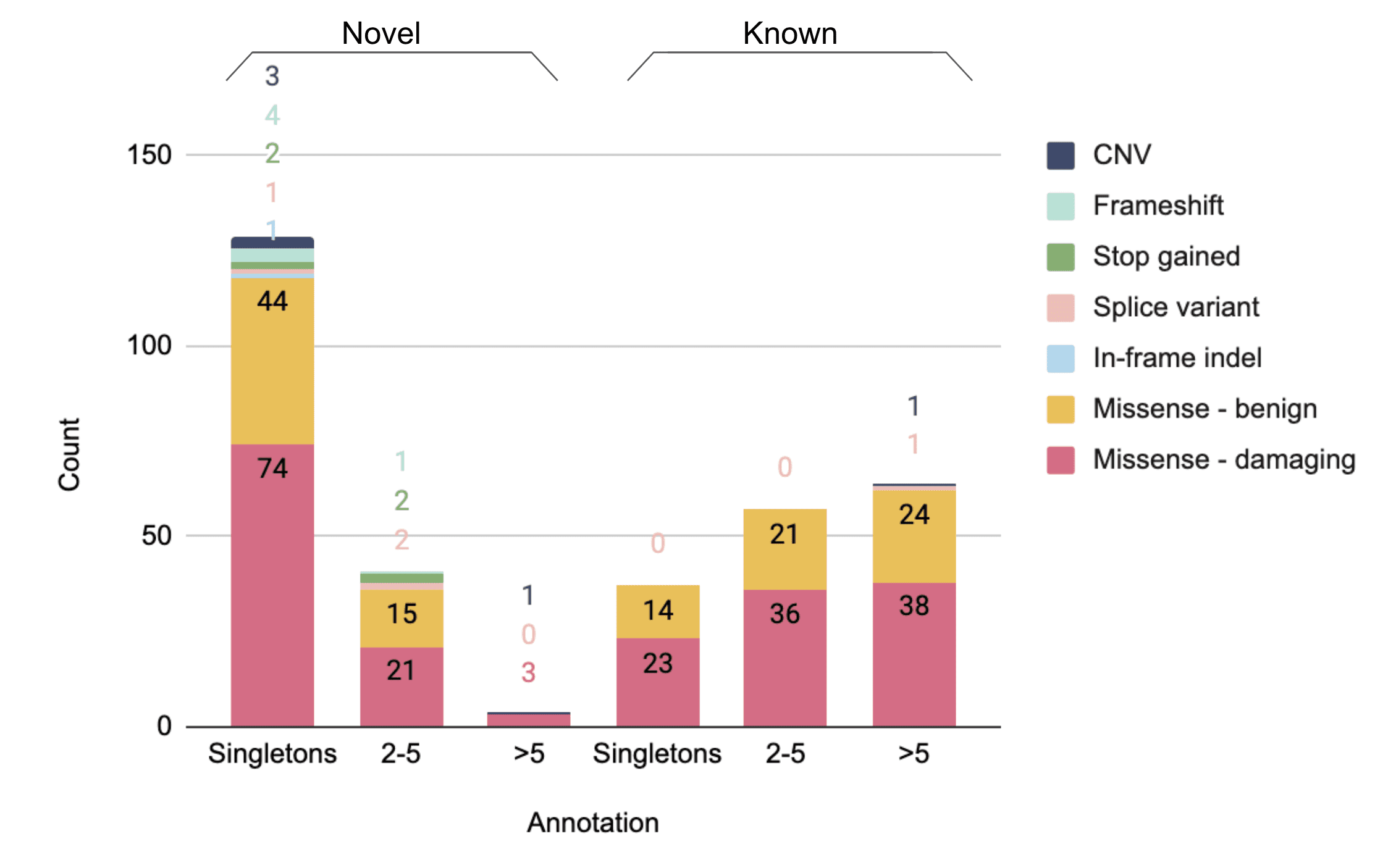
Figure 1. ACE2 variants found in the Helix dataset of >200,000 unrelated individuals. Count of coding variants by type, grouped by singleton, 2-5 carriers, or >5 carriers and whether novel or in gnomAD database(s).
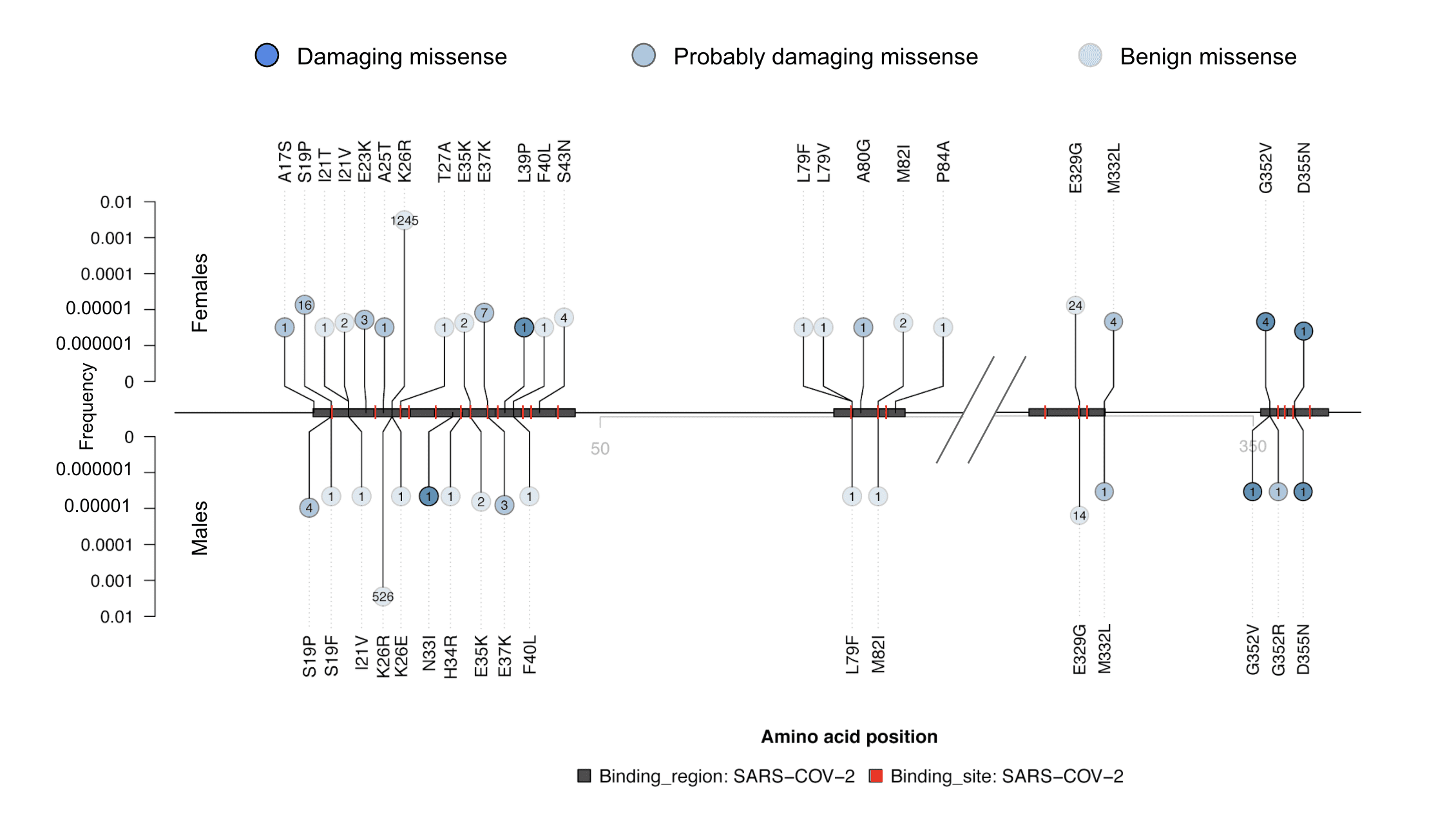
Figure 2. Lolliplot10 of missense variants in the regions of ACE2 that bind to SARS-CoV-2. The amino acid positions along ACE2 are shown along the horizontal middle line, SARS-CoV-2 binding regions8,9 are shown as gray boxes, and the specific amino acids that bind to SARS-CoV-2 are highlighted in red. For clarity, only the binding regions are depicted (0-95 and 315-360 of 805 amino acids). The vertical line height above the middle line represents allele frequencies in females, and below are allele frequencies in males (log scale). The missense variants in these regions are annotated as damaging (SIFT <0.05 and Polyphen2 >0.85; dark blue), probably damaging (SIFT <0.05 or Polyphen2 >0.15; medium blue), or benign (SIFT >0.05 and Polyphen2 <0.15; light blue). All coding variants in the Helix dataset that overlap the binding regions are shown, with case counts of each variant indicated in the bubble, and amino acid change indicated.
Currently, the human host receptor for this virus is thought to be Angiotensin I converting enzyme 2 (ACE2)5,6, a 19-exon gene encoding a protein of 805 amino acids, with only one major protein-coding isoform. ACE2 is a carboxypeptidase transmembrane protein that cleaves angiotensin I and II, controls vasoconstriction and blood pressure (and therefore hypertension), and was also previously established as the host receptor for SARS-CoV (from the 2002 epidemic). It has high expression levels in multiple tissues, especially those of the small intestine and testis. Multiple common variants in linkage disequilibrium (LD) with each other are already known to be associated with ACE2 expression levels in the brain, but not in other tissues, especially those with high expression of the gene. Importantly, ACE2 is on the X chromosome. Because males have only one copy of the X chromosome and females have two, this means that any males carrying one copy of a variant in this gene are more likely to see its effect on their phenotype than would a female carrying one copy of the same variant. Finally, mouse models with a presumed knockout of the ACE2 gene showed cardiac, metabolic, muscle and pulmonary phenotypes, including pulmonary vascular congestion and increased lung weight, and they were more likely to die early of congestive heart failure after transverse aortic constriction.
At Helix, we queried our database of more than 200,000 exomes of unrelated people to identify the frequencies of different coding variants, including CNVs, in ACE2. We have identified 332 variants that affect the coding sequence of this gene, 16 of which are loss of function (LoF), five of which are CNVs, and 174 of which are not found in either gnomAD v2.1.1 (115,000 exomes and 15,000 genomes) or gnomAD v3 (70,000 genomes) (Figure 1)7. The frequencies of these variants in unrelated individuals from populations of different ancestries can be found here. Importantly, we compared the variants we found to the recently reported structure of ACE2 bound to SARS-CoV-28,9. We identified 11 coding variants in 83 individuals that changed the specific amino acids shown to physically interact with SARS-CoV-2, and an additional 29 variants in 1,885 individuals that were within two amino acids of these crucial sites (Figure 2). The most common among these variants is chrX:15600835:T:C / p.K26R, which has an allele frequency of ~0.5% in multiple populations and changes the amino acid directly adjacent to a SARS-CoV-2-interacting amino acid but is not predicted to be damaging. Of the 40 variants in the binding regions, 13 were predicted to be damaging, and 3 of these were specifically on the interactive amino acids. Notably, one of the damaging variants that directly affects a SARS-CoV-2 interacting amino acid, chrX:15600857:A:G / p.S19P, is very rare or absent in most populations but has an allele frequency of 0.1-0.2% in those with African ancestry. Variants in these interacting regions may change the strength of the bond with SARS-CoV-2, which could have effects ranging from being protective to increasing risk, and they are interesting genomic targets for further study.
Of these variants, we identified only a single individual with an ACE2 full-gene deletion. As it turns out, the individual was female, and retained one functioning copy of ACE2 on their other X chromosome. However, if a male had the gene deleted, then they would be entirely missing this gene. Likewise, other LoF variants would result in no working copies of ACE2 in a male but might have no effect in a female. Looking at >200,000 individuals sequenced at Helix, we find that predicted LoF variants collectively have an allele frequency of 0.013% in females but 0.007% in males for those of European ancestry, and <0.05% in non-European ancestry females and males (Figure 3). Among high-confidence LoF variants as annotated by LOFTEE, the frequencies were 0.01% in European ancestry females and 0.005% in males. No females were homozygous or compound heterozygous for LoF variants. Additionally, we observe no frameshift variants in males despite finding them in six females (Figure 4); most of the LoF variants seen in males are splice variants, and only a single male was found to have a premature stop codon, in the C-terminal collectrin domain. This pattern is consistent with hemizygous depletion of LoF variants in this gene (Fisher’s exact p=0.002), meaning that the complete loss of this gene is likely deleterious or even lethal. Furthermore, this result is consistent with the data available from gnomAD’s ~185,000 exomes and genomes, where the LoF allele frequency is 0.009% in females of European ancestry as compared to 0.003% in males (non-European ancestries, <0.02% in females and males) and the UK Biobank’s ~50,000 exomes, where it is 0.013% in females of British ancestry and 0.006% in males, and by its published probability of being loss-of-function intolerant (pLI) score of 111. Because LoF variants are so rare in this gene, the exact frequency in specific non-European ancestry populations, which have smaller sample sizes available in these large databases, cannot be precisely calculated.
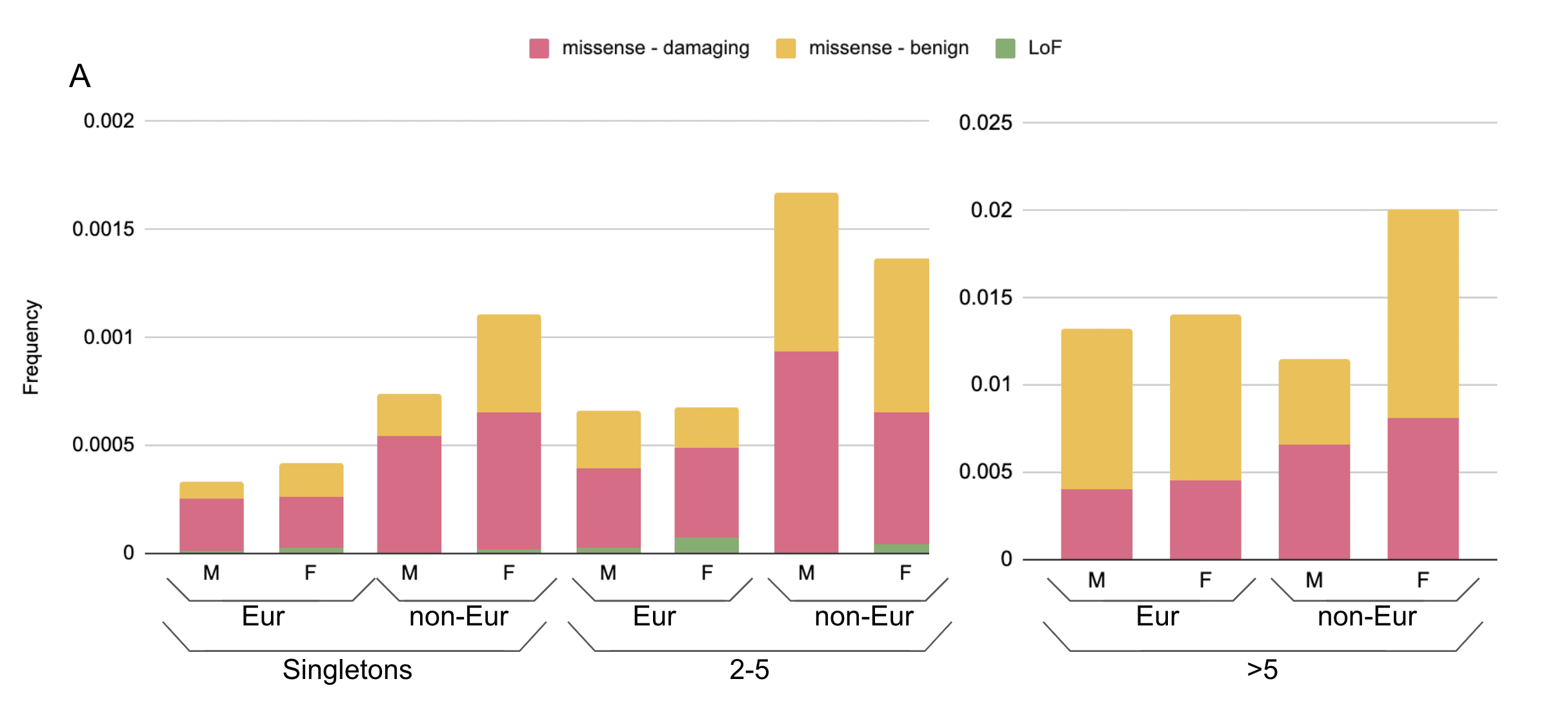
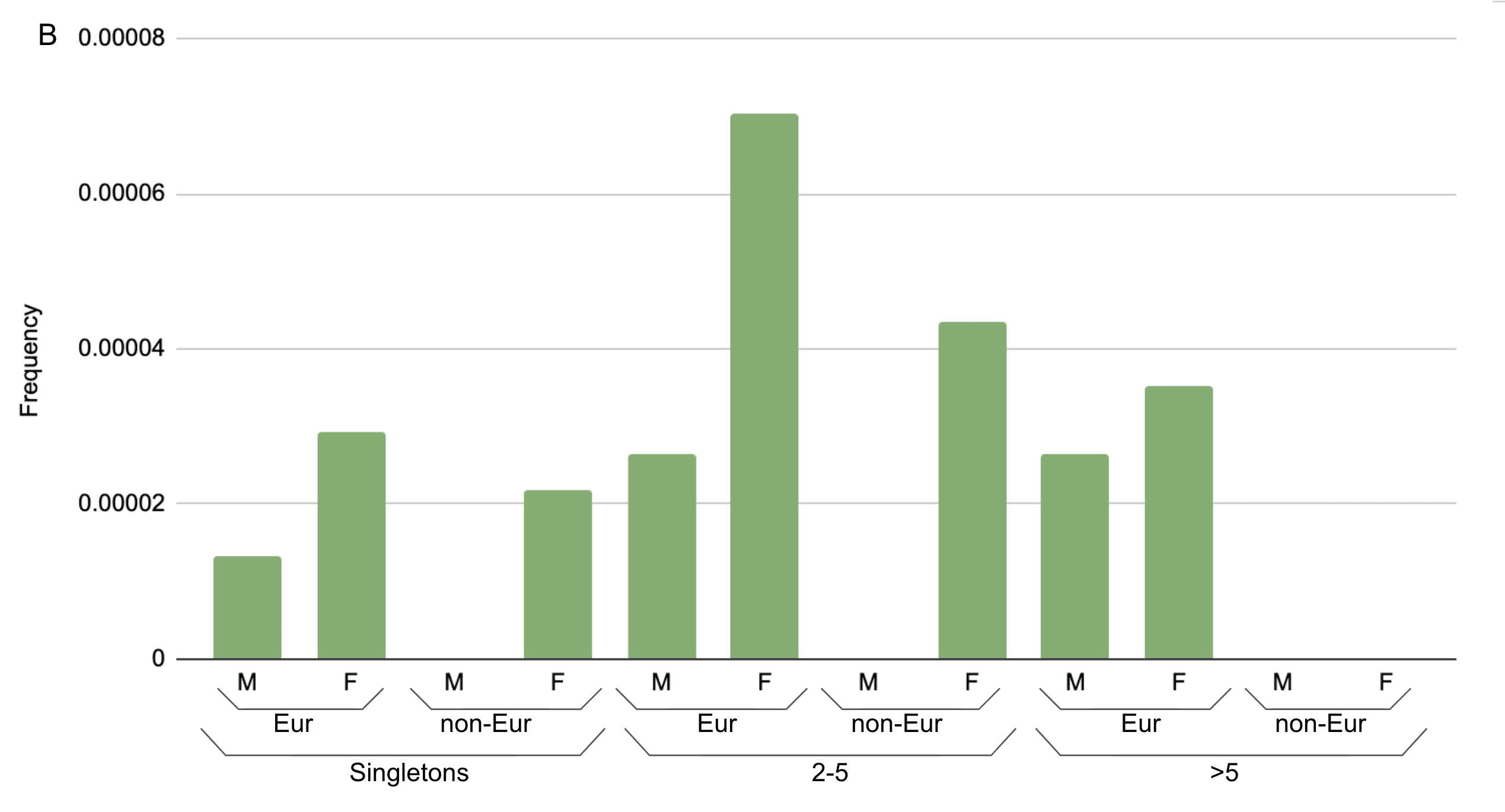
Figure 3. Aggregated frequency of variants found in the Helix dataset of >200,000 unrelated individuals. A) Proportion of individuals in Helix dataset of European and other (non-Eur) ancestries carrying coding variants; (B) carrying only LoF variants. Frequencies are broken up according to the number of carriers seen in the Helix dataset (singletons, 2-5 carriers, or >5 carriers). M=male, F=female.
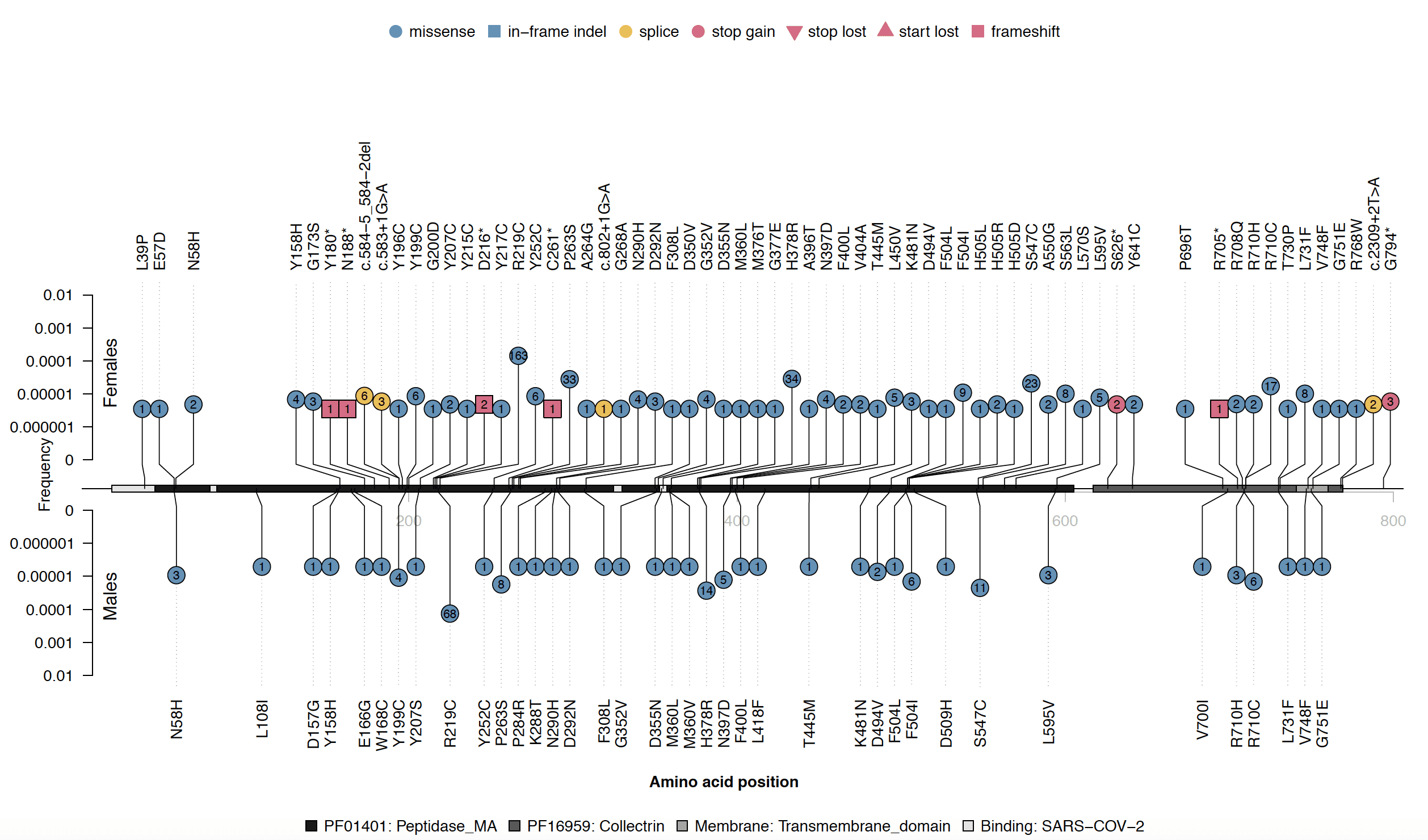
Figure 4. Lolliplot10 of damaging coding variants found in ACE2. Variants shown are from the unrelated European ancestry individuals in the Helix dataset. The amino acid positions along ACE2 are shown at the horizontal middle line together with the different PFAM domains, the C-terminal transmembrane domain, and the SARS-CoV-2 binding regions8 shown as gray boxes. Vertical lines represent variant frequencies (log scale) in females (above) and males (below). The type of variant is indicated by the shape and color of each lollipop, as well as the number of carriers within each. Variants were defined as damaging according to a consensus between SIFT and Polyphen2 (as in Figure 2). Y axis is log scale.
Of the CNVs identified in our Helix dataset, besides the whole-gene deletion in one individual noted above, we found two individuals who also had a single exon deleted each and 0.02% of our population who had a duplication of the first 6-7 exons of the gene, each of which also has the potential to cause loss of function. All of these CNVs were found in females. The only CNV that we found in males was a duplication of the entire ACE2 gene, which is duplicated in ~0.04% of our population. This is consistent with the frequency of this duplication shown in gnomAD. As a whole-gene duplication could increase expression, we compared the genotypes in our dataset to those in the GTEx database. In GTEx, there is a very common haplotype associated with differential expression of the gene in some tissues, extending from the 5’-end of the gene to far upstream and into the next gene, an ACE2 homolog with 45% identity to ACE2 but no catalytic domain12. We found that in the Helix dataset, the carriers of the rare whole-gene duplication were on the very common low-expression ACE2 haplotype. Although this does not mean that duplication carriers have low expression of the gene, it does show that the carriers of this rare duplication variant are not the drivers of the association between expression and that haplotype, as does the spread-out nature of the common variant’s violin plot (Figure 5).
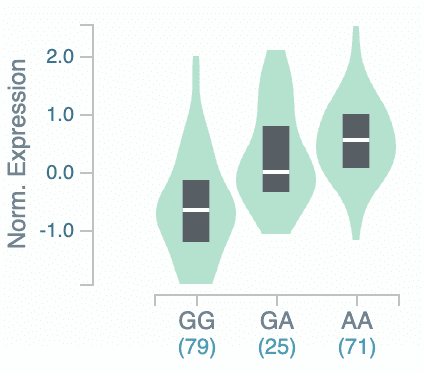
Figure 5. Association between ACE2 variant rs4830974 and Brain Frontal Cortex (BA9). Exemplar genotype for low-expression eQTL from GTEx, p=2.6×10-17. All other eQTL in LD with this genotype had similar expression levels.
Finally, we looked into non-COVID-19-related phenotypes that might be associated with variation in ACE2. In our recent analysis of rare coding variants in the Healthy Nevada Project and UK Biobank13–15, we did not identify any genome-wide significant associations between the >4,000 phenotypes analyzed and the collapsed rare coding variants in ACE2. However, we highlight here the top 20 associations from the meta-analysis across the two cohorts.
Top 20 identified ACE2 associations ▼
| P-value | Direction | Model | Method | Case Carriers | Ctrl Carriers | Phenotype |
|---|---|---|---|---|---|---|
| 0.001 | –– | Coding | LMM | 128 (0.0026%) | N/A | Basophill percentage |
| 0.004 | +0 | Coding | FET | 3 (0.0306%) | 129 (0.0026%) | G99 Other disorders of nervous system in diseases classified elsewhere |
| 0.004 | ++ | Coding | FET | 5 (0.013%) | 127 (0.0026%) | W10 Fall on and from stairs and steps |
| 0.005 | +0 | Coding | FET | 3 (0.0306%) | 129 (0.0026%) | H35.8 Other specified retinal disorders |
| 0.005 | ++ | Coding | FET | 5 (0.0124%) | 127 (0.0026%) | M47.9 Spondylosis, unspecified |
| 0.006 | -- | LoF | LMM | 7 (0.0001%) | N/A | Basophill percentage |
| 0.007 | -- | Coding | LMM | 19 (0.0015%) | 113 (0.0031%) | I10 Essential (primary) hypertension |
| 0.009 | ++ | Coding | FET | 10 (0.0065%) | 122 (0.0026%) | K21.0 Gastro-oesophageal reflux disease with oesophagitis |
| 0.011 | ++ | LoF | LMM | 7 (0.0001%) | N/A | Standing height |
| 0.011 | ++ | Coding | LMM | 23 (0.0039%) | 109 (0.0025%) | K21 Gastro-oesophageal reflux disease |
| 0.013 | -- | Coding | LMM | 10 (0.0012%) | 147 (0.0023%) | R07 Pain in throat and chest |
| 0.014 | ++ | Coding | FET | 2 (0.0317%) | 130 (0.0027%) | B96.1 Klebsiella pneumoniae as the cause of diseases classified to other chapters |
| 0.014 | +0 | Coding | FET | 6 (0.0086%) | 126 (0.0026%) | Y83.1 Surgical operation with implant of artificial internal device |
| 0.015 | ++ | Coding | FET | 5 (0.0092%) | 127 (0.0026%) | M15.9 Polyarthrosis, unspecified |
| 0.015 | ++ | Coding | LMM | 16 (0.004%) | 141 (0.0021%) | R10.1 Pain localised to upper abdomen |
| 0.016 | +0 | Coding | FET | 2 (0.04%) | 130 (0.0027%) | S82.3 Fracture of lower end of tibia |
| 0.021 | -- | Coding | LMM | 130 (0.0026%) | N/A | Platelet count |
| 0.023 | 0+ | Coding | FET | 2 (0.0308%) | 130 (0.0027%) | I67.1 Cerebral aneurysm, nonruptured |
| 0.023 | -- | Coding | LMM | 73 (0.0023%) | N/A | Potassium |
| 0.024 | ++ | Coding | FET | 4 (0.008%) | 128 (0.0026%) | K63.8 Other specified diseases of intestine |
LMM = linear mixed model; FET = Fisher’s exact test; ++ = positive direction in both cohorts; – – =negative association in both cohorts; +0 = no case carriers in HNP cohort; 0+ = no case carriers in UK Biobank cohort. For quantitative traits, the total number of carriers with phenotypes measured is shown in the Case column, and N/A in the control column. P-values are uncorrected for multiple tests.
Consistent signals were seen in both cohorts for a weak association between those carrying coding variants in ACE2 and having lower basophil percentages. Some of the other signals observed included a weak protective effect of coding variants for hypertension, which is interesting given this gene’s function in blood pressure and the known relationship between the related gene ACE and human blood pressure16; and a predisposition for those with coding variants to have gastro-oesophageal reflux disease with oesophagitis, which is consistent with the nominal association observed between the low-frequency intronic variant rs183546232 in ACE2 (MAF 1%) and eosinophilic esophagitis in an independent dataset. There was also a nominal association between coding variants and ICD10 code B96.1, Klebsiella pneumoniae as the cause of diseases classified elsewhere, but this was driven by only 2 case carriers across both cohorts (although Fisher’s exact test is robust to small samples). Investigations of additional pneumonia-related ICD10 codes such as J18 revealed no significant association with ACE2 variation.
We also utilized the existing publicly available UK Biobank GWAS results from the Neale lab to perform a pheWAS of common variants in ACE2. We did not identify individual common variants in this gene with significant associations with any phenotypes. We anticipate, with the sizable global scientific efforts dedicated to COVID-19 host genomics, that potential associations between ACE2 variants and COVID-19 response will be characterized once human genetic data paired with COVID-19 phenotypes are generated.
In conclusion, there is moderate evidence that ACE2 variation is associated with gastro-oesophageal reflux disease with oesophagitis, and limited evidence of it having a strong role in other phenotypes in the absence of COVID-19. Homo/hemizygous LoF of ACE2 appears to be infrequently tolerated and is likely extremely deleterious. Although LoF variants in ACE2 are extremely rare, variants affecting the regions that interact with SARS-CoV-2 are more prevalent, and the impending research studies on human genetics and COVID-19 response may identify some of these variants as conferring resistance or heightened susceptibility to the virus.
References
1. Kaiser, J. How sick will the coronavirus make you? The answer may be in your genes. Science (2020) doi:10.1126/science.abb9192.
2. Liu, R. et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367–377 (1996).
3. Samson, M. et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382, 722–725 (1996).
4. Dean, M. et al. Genetic Restriction of HIV-1 Infection and Progression to AIDS by a Deletion Allele of the CKR5 Structural Gene. Science 273, 1856–1862 (1996).
5. Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
6. Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 94, (2020).
7. Karczewski, K. J. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 531210 (2019) doi:10.1101/531210.
8. Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature (2020) doi:10.1038/s41586-020-2179-y.
9. Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
10. Ou, J. & Zhu, L. J. trackViewer: a Bioconductor package for interactive and integrative visualization of multi-omics data. Nat. Methods 16, 453–454 (2019).
11. Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
12. Chu, P.-L. & Le, T. H. Role of collectrin, an ACE2 homologue, in blood pressure homeostasis. Curr. Hypertens. Rep. 16, 490 (2014).
13. Cirulli, E. T. et al. Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts. Nat. Commun. 11, 542 (2020).
14. Researchers have access to new data on thousands of exomes. Here’s what we found. – Helix. Helix https://blog.helix.com/uk-biobank-helix-research/ (2019).
15. We’ve analyzed over 1,000 more phenotypes using the UK Biobank’s latest data – Helix. Helix https://blog.helix.com/uk-biobank-helix-research-blood-biomarkers/ (2019).
16. Wain, L. V. et al. Novel Blood Pressure Locus and Gene Discovery Using Genome-Wide Association Study and Expression Data Sets From Blood and the Kidney. Hypertension (2017) doi:10.1161/HYPERTENSIONAHA.117.09438.